1。遺伝的マッピング:
* リンケージ分析: この古典的な手法は、関心の特性(病気など)とともに、遺伝マーカー(SNPやマイクロサテライトなど)の遺伝パターンを分析します。マーカーと特性が一緒に継承される傾向がある場合、それらは染色体に近い可能性があります。 この方法は、多くの遺伝性疾患の遺伝子のマッピングに役立ちました。
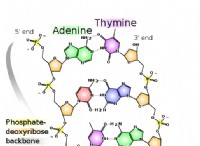
* 再結合頻度: 減数分裂中の組換えイベントは、染色体間の遺伝物質を交換します。 2つのマーカー間の組換えの頻度は、それらの間の距離に比例します。これにより、遺伝子間の相対距離を示す遺伝子マップの作成に役立ちます。
2。物理マッピング:
* 染色体バンディング: 染色体は染料で染色され、特徴的なバンディングパターンを明らかにします。これらのパターンは一意であり、特定の染色体領域の同定と局在化を可能にします。
* 蛍光in situハイブリダイゼーション(FISH): 蛍光標識DNAプローブは、染色体の特定の領域にハイブリダイズするために使用されます。蛍光信号は、標的DNA配列の位置を明らかにします。
* クローンライブラリ: ヒトゲノム全体を表すDNAフラグメントは、ライブラリにクローン化され、保存されます。これらのライブラリは、特定の遺伝子またはDNA領域のプローブを使用してスクリーニングでき、物理マッピングを可能にします。
3。次世代シーケンス(NGS):
* 全ゲノムシーケンス: NGSは、ヒトゲノム全体をシーケンスすることができ、遺伝子の同定と局在のための完全な青写真を提供します。
* exomeシーケンス: これは、ゲノムのタンパク質コーディング領域(エクソン)のみを配列決定することに焦点を当てており、これは疾患原因となる遺伝子の特定に特に関連しています。
* ゲノムワイド関連研究(GWAS): このアプローチは、特定の特性または疾患を持つゲノム全体にわたるDNA配列(SNP)の変動間の関連を分析します。 GWASは、複雑な特性や疾患に関与する遺伝子を特定するのに役立ちます。
4。データベースとオンラインリソース:
* ncbi遺伝子: このデータベースは、染色体の位置、配列、機能、および関連する疾患を含む、ヒト遺伝子に関する包括的な情報を提供します。
* UCSCゲノムブラウザ: このインタラクティブなブラウザにより、ユーザーは遺伝子注釈、バリエーション、その他のゲノム機能など、ヒトゲノムを視覚化および分析できます。
5。計算アプローチ:
* バイオインフォマティクス: 計算アルゴリズムとツールは、膨大な量のゲノムデータを分析し、遺伝子位置を予測し、疾患の潜在的な候補遺伝子を特定するために使用されます。
重要な考慮事項:
* 遺伝的変異: 人間は高度な遺伝的変異を持っているため、遺伝子の位置を正確に特定し、特性や疾患に関連する一般的なバリアントを特定するために、大きな集団を研究することが重要です。
* 遺伝子調節: 遺伝子の位置は、遺伝子機能を決定する唯一の要因ではありません。 プロモーターやエンハンサーなどの調節要素は、遺伝子発現の制御に重要な役割を果たします。
* エピジェネティクス: 環境要因は、DNA配列を変更せずに遺伝子発現に影響を与える可能性があります。これらのエピジェネティックな修正を理解することは、健康と病気における遺伝子機能を理解するために重要です。
これらの多様なアプローチを組み合わせることにより、科学者は人間のゲノムの理解を絶えず改善し、遺伝子機能と健康と病気におけるその役割についての新しい発見につながります。